Cloudgene 3¶
Turn Your Nextflow Pipeline into a Powerful Web Service¶
Getting Started Installation Source
Documentation of Cloudgene 2
Build your analysis pipeline in Nextflow
Integrate your analysis pipeline into Cloudgene by writing a simple configuration file
Get a powerful web application with user management, data transfer, error handling and more
Deploy your application with one click to in-house clusters or public Clouds like Amazon AWS
Offer your application as SaaS to other scientists, managing thousands of jobs
Share your application, enabling others to clone your service to their own hardware or private cloud instance
Integrate Your Nextflow pipelines¶
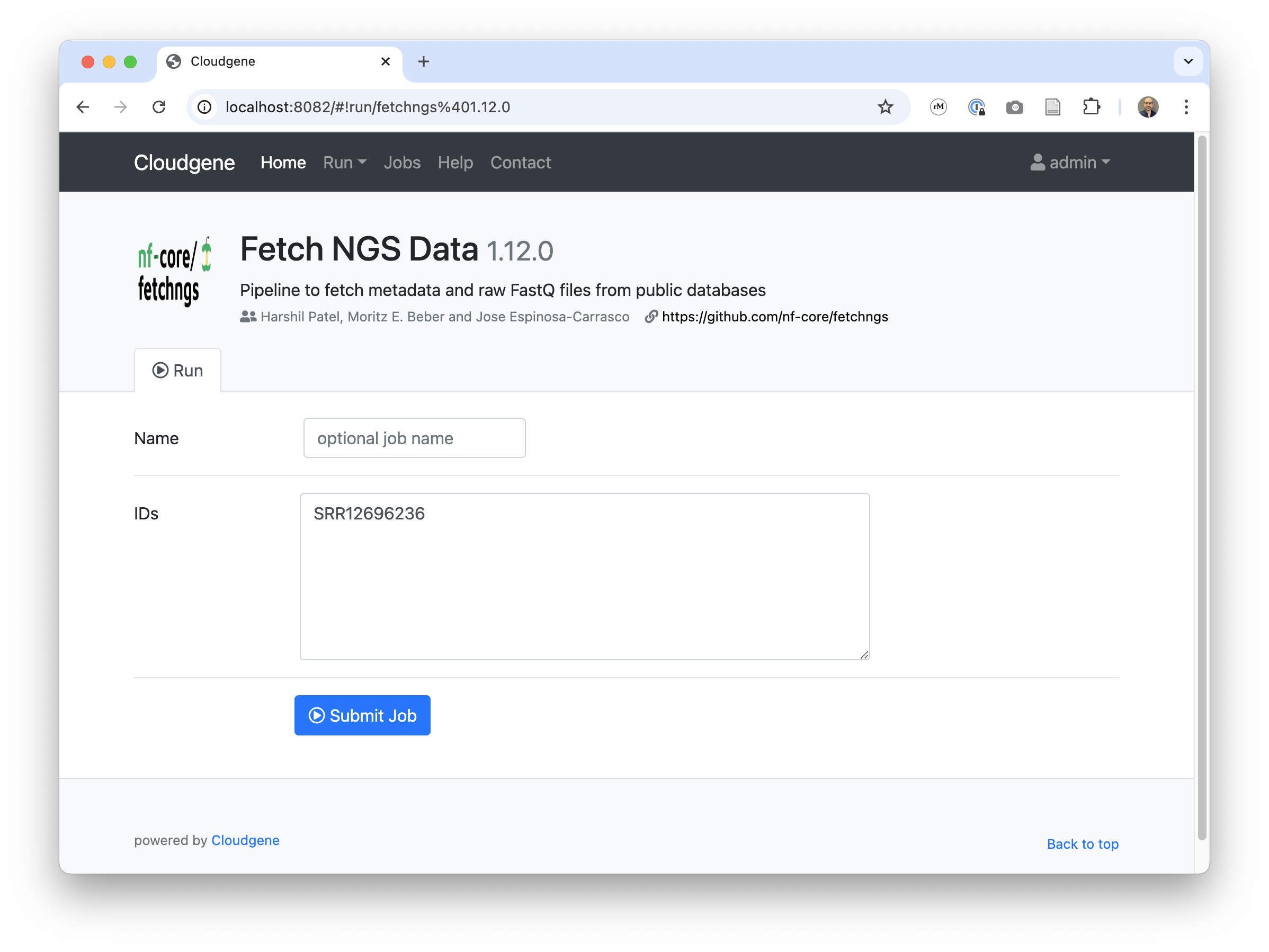
Integrate your analysis pipeline into Cloudgene by writing a simple configuration file.
id: fetch-ngs
name: FetchNGS
description: Pipeline to fetch metadata and raw FastQ files from public databases
version: 1.12.0
website: https://github.com/nf-core/fetchngs
workflow:
steps:
- name: Fetch NGS
script: nf-core/fetchngs
revision: 1.12.0
inputs:
- id: input
description: IDs
type: textarea
value: "SRR12696236"
writeFile: "ids.csv"
outputs:
- id: outdir
description: Output
type: folder
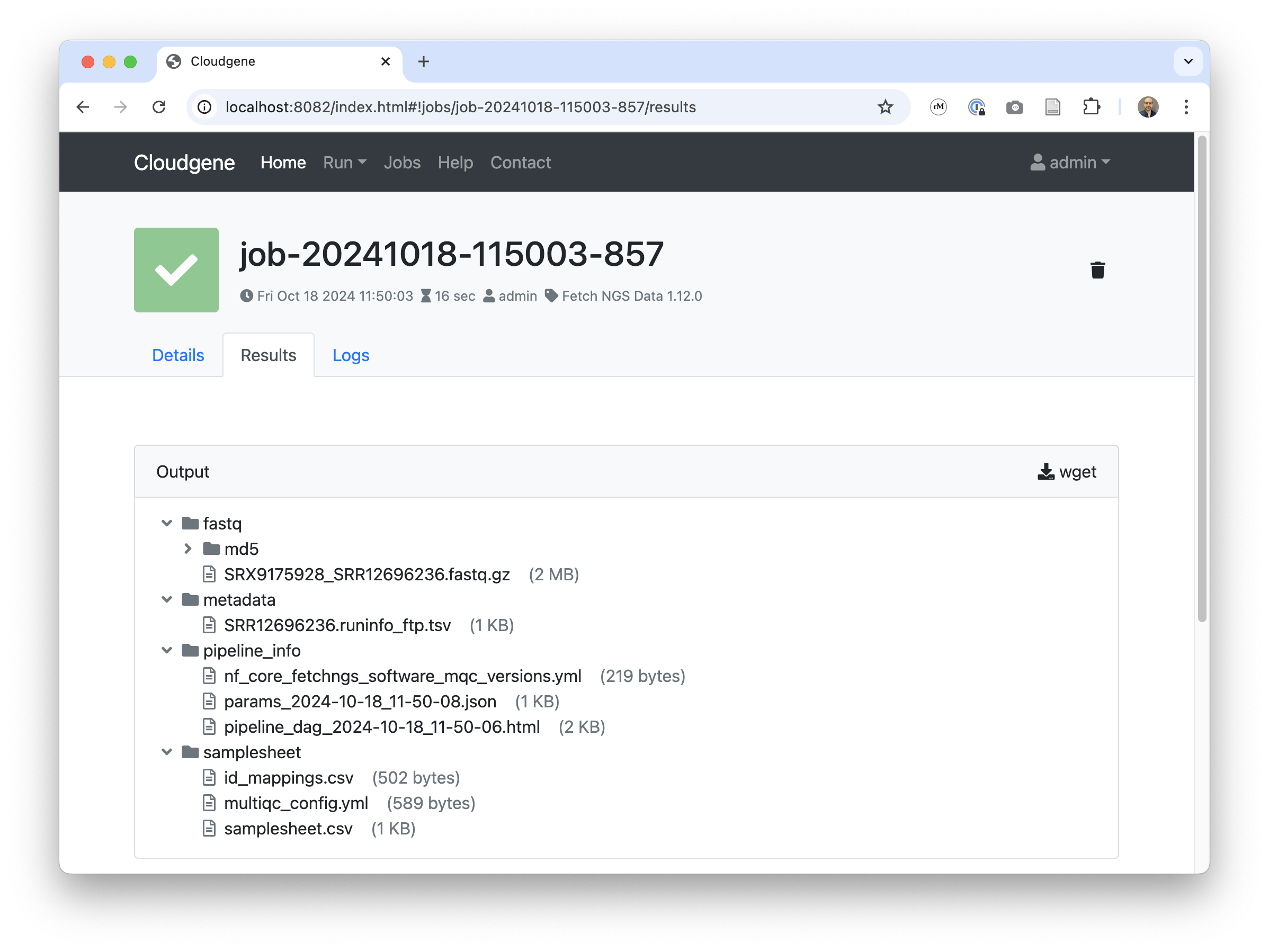
Share your pipeline¶
Share your application via HTTP, GitHub, or S3, and enable users to install it with a simple command.
Combine Your Nextflow pipeline with others¶
Combine your Nextflow pipeline with other pipelines and create use-case specific web services.
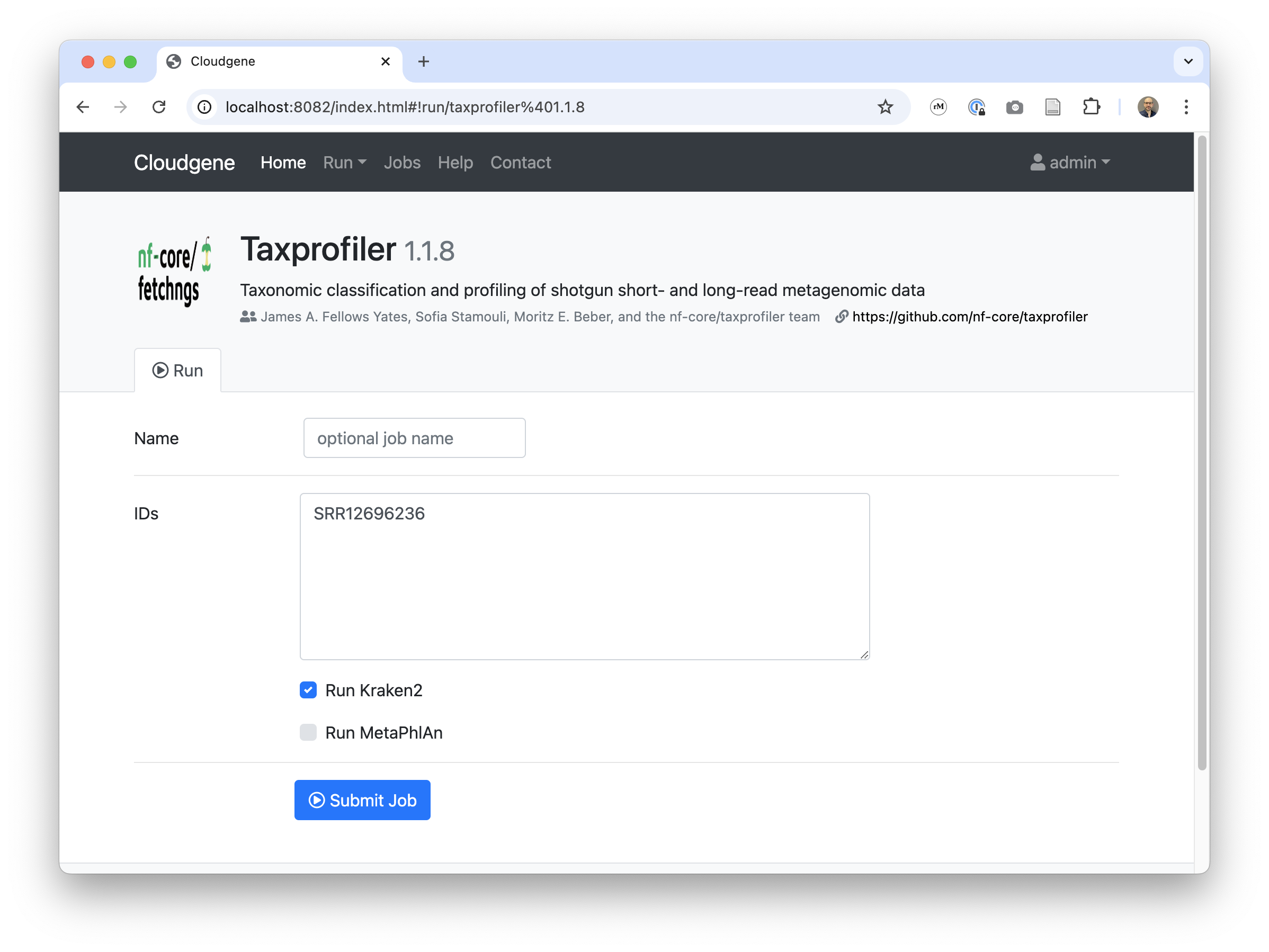
id: taxprofiler
name: Taxprofiler
description: Taxonomic classification and profiling of shotgun short- and long-read metagenomic data
version: 1.1.8
website: https://github.com/nf-core/taxprofiler
author: James A. Fellows Yates, Sofia Stamouli, Moritz E. Beber, and the nf-core/taxprofiler team
logo: https://raw.githubusercontent.com/nf-core/fetchngs/master/docs/images/nf-core-fetchngs_logo_light.png
workflow:
steps:
- name: Fetch Data
script: nf-core/fetchngs
revision: 1.12.0
stdout: true
params:
input: "${input_ids}"
- name: Run taxprofiler
script: nf-core/taxprofiler
revision: 1.1.8
stdout: true
params:
input: "${outdir}/samplesheet/samplesheet.csv"
databases: "https://raw.githubusercontent.com/nf-core/test-datasets/taxprofiler/database_full_v1.2.csv"
multiqc_title: "${CLOUDGENE_JOB_NAME}"
inputs:
- id: input_ids
description: IDs
type: textarea
value: "SRR12696236"
writeFile: "ids.csv"
serialize: false
outputs:
- id: outdir
description: Output
type: folder

Who uses Cloudgene?¶
Michigan Imputation Server 2¶
Michigan Imputation Server 2 provides a free genotype imputation service (chromosomes 1-22, chromosome X and HLA region) using Minimac4. You can upload phased or unphased GWAS genotypes and receive phased and imputed genomes in return. Our server supports imputation from numerous reference panels. For all uploaded datasets a comprehensive QC is performed. The complete source code is available on GitHub.
mtDNA-Server 2¶
mtDNA-Server 2 is a Nextflow DSL2 pipeline to accurately detect heteroplasmic and homoplasmic variants in mitochondrial (mtDNA) genomes. The complete source code can be acccessed here.
Haplocheck¶
Haplocheck detects in-sample contamination in mtDNA or WGS sequencing studies by analyzing the mitchondrial content.
Citation¶
Cloudgene 3: Transforming Nextflow Pipelines into Powerful Web Services. Forer L, Schoenherr S. bioRxiv 2024.10.27.620456; doi: https://doi.org/10.1101/2024.10.27.620456
About¶
Cloudgene has been created by Lukas Forer and Sebastian Schönherr and is MIT Licensed.


Thanks to all the contributors to help us maintaining and improving Cloudgene!